For life science service or technology providers engaged in clinical research, it’s imperative to ensure compliance with international standards. Even though the ultimate responsibility for compliance lies with the sponsor, you are also required to adhere to the rules enforced by the sponsor. This blog post outlines seven key responsibilities for compliance with an important and widely recognized ethical standard: ICH-GCP E6.
What is ICH-GCP E6?
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is an important initiative that brings together regulatory authorities around the world.
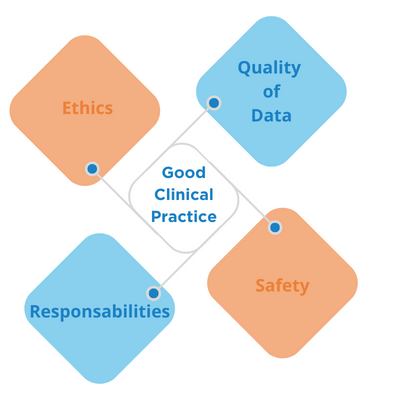
ICH-GCP E6 (Good Clinical Practice) serves as the international ethical and scientific quality standards for designing, conducting, recording, and reporting trials involving the participation of human subjects. It ensures that the rights, safety, and well-being of trial subjects are protected.
The protection of clinical trial subjects is consistent with the principles set out in the Declaration of Helsinki (ethical principles for medical research involving human subjects, October 2013).
Ensuring compliance with ICH-GCP E6
Adherence with ICH-GCP E6 provides public assurance that the:
- Rights, safety, and well-being of trial subjects are protected.
- The clinical trial data are credible and well documented.
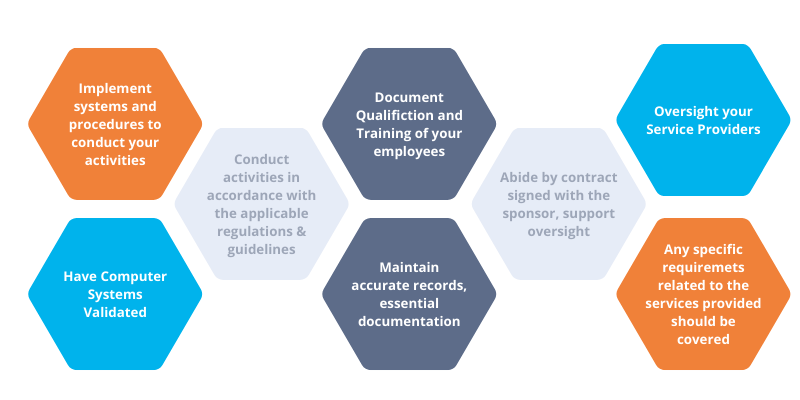
7 ICH-GCP responsibilities for life science service providers
If you’re starting your compliance journey, here are some key elements to help you understand your responsibilities and establish measures to align with the current version of ICH-GCP.
Note: This is a non-exhaustive list, and provided SOP titles are presented as examples to guide you.
As a Contract Service Organization (CRO) or service provider, you are responsible for implementing and maintaining a quality management system (QMS) from day one in compliance with the requirements of the ICH GCP E6 standard.
- Implementation of a Quality Management System
The QMS may be paper-based or software-managed. Either way, the (e)QMS should encompass policies about:
- Quality
- Data integrity
- IT security
- Confidentiality
The (e)QMS should also outline Standard Operating Procedures (SOPs) covering all aspects of activities related to Sponsor projects and internal process, such as Quality Assurance, Quality Control, Computer System Validation.
At the end of collaboration with a sponsor, the list of applicable QMS documentation might be requested.
2. Conducting business in accordance with regulations
To guarantee compliance with ICH-GCP and the applicable regulatory requirement(s), your company should develop an SOP on Regulatory Compliance.
This SOP should outline activities and responsibilities within your company for managing updates on regulatory requirements, ensuring regulatory intelligence is integrated into your operations.
3. Documentation of employee qualification and training
Any of your employees involved in the conduct of a clinical trial must be competent to perform their duties and qualified by education, training, and experience. Staff performing work affecting clinical research quality or customer project quality shall be competent based on appropriate education, training, skills, and experience in compliance with appropriate regulations, guidelines, and your policies and SOPs.
Personnel qualification and training should be managed according to SOPs. For example:
- SOP-Personnel Qualification demonstrates that everyone employed by your company is qualified prior to independently perform their assigned function, duty, or task as established by their job requirements (each employee should have an up-to-date job description).
- SOP-Personnel Training describes the process for preparing, providing, and maintaining your staffs’ trainings to demonstrate their appropriate qualification to conduct activities.
ICH-GCP training should be documented and referenced in the CV, with a refresher training recommended every 2 years.
4. Documentation of sponsor delegation and support sponsor oversight
Any trial-related duty and function that is transferred to and assumed by your company should always be specified in writing, usually through a Master Service Agreement and Work Order.
To support any Sponsor regarding its oversight responsibility of trial-related duties that are subcontracted to you:
- Define roles and responsibilities in the Work order.
- Propose that the Sponsor review/approve essential specific documentation.
- Provide regular updates on the project activities (to be defined upfront).
- Provide a list of SOPs and CVs.
- Host a sponsor’s audit (e.g., during which computer system validation documentation and (e)QMS documentation are accessible).
5. Management of your CROs
Any trial-related duty and function that your company transfers or subcontracts to another CRO should be specified in writing. This can be covered by SOP-Contractual Agreement Management with CRO/service provider.
Before initiating any activities, you should have a SOP in place that defines the process to manage your providers to ensure they can demonstrate the mandatory qualification, experience, facilities, equipment, and quality systems to perform subcontracted activities in compliance with Good Clinical Practice standard (ICH-GCP E6) or any other regulatory requirements (e.g., General Data Protection Regulation (GDPR)).
You should maintain the list of your qualified CROs/services providers.
6. Validation of your computer systems
For companies developing or using computer systems, it is important to ensure that computer systems are built, supported, and maintained in compliance with applicable regulations during their entire life cycle. The computer systems should fit for their intended use and comply with applicable regulations (e.g., 21 CFR part 11).
The validation process for the computer systems should be described in a set of SOPs for:
- Computer System Validation.
- Computer System Testing.
- Computer System Risk Assessment.
Any change to the system should follow a formal process to keep the system qualified. This process should be described in SOP-IT Change Management.
You may also have SOPs in place covering data backup and recovery.
7. Maintenance of accurate records (e.g., Essential Documentation)
Retain all essential sponsor-specific documents relating to your activities in the trial in accordance with the applicable regulatory requirements of the country(ies) where the product is approved and/or where the sponsor intends to seek approval(s).
The documents generated during a sponsor project conduct should be timely filed in a Trial Master File (TMF), centralizing project management documentation. This TMF can be paper based or electronic.
About ICH E6(R3)
Good clinical practice, like any subset of GxP, is constantly evolving.
On 19 May 2023, the ICH released a long-awaited draft guideline, ICH E6 (R3), which addresses evolving trial designs and technological innovations for risk-based approaches.
While working on implementing ICH E6 (R2), you should take a look at how R2 differs from R3.
ICH E6(R3) has been restructured and is composed of an overarching principles section, Annex 1 (interventional clinical trials), Annex 2 (additional considerations for non-traditional interventional clinical trials), Glossary, and Appendices.
The overarching principles, Annex 1, Glossary, and Appendices will replace the current E6(R2).
A new section titled ‘Data Governance’ includes new and expanded text on data capture, data verification, metadata, data corrections, data transfers, and finalization of datasets prior to analysis.
Conclusion
While ICH-GCP compliance is vital, you should identify additional Regulations & Principles your company is bound to. For example:
- 21 CFR Part 11,
- EU Annex 11,
- General Data Protection Regulation (GDPR),
- HIPPA.
Finally, in parallel to implementing GCP, bear in mind that the new GCP guideline is a significant revision that includes new content. The impending of draft guideline ICH E6(R3) underscores the importance of staying informed and preparing for changes in electronic systems, data management, and data integrity.
Proactive familiarization is key to ensuring seamless compliance when final Annex 2 will be released in 2024. As a life science service provider or technology company, these steps will not only enhance your GCP compliance but also establish a robust foundation for successful clinical research endeavors.